
Positive: Responsiveness , Value Thank you Parker Waichman and a special thanks to Francisco for successfully settling such a complex case like mine. Would refer a friend.
Chulani Sterling
3 years ago
Voluntary worldwide recall of 1,184 PIC50 External Monitor/Defibrillators. MRL, Inc., a Welch Allyn Company, today announced it is initiating a voluntary worldwide recall of 1,184 PIC50 External Monitor/Defibrillators manufactured in Buffalo Grove, IL between February 2002 and October of 2004. These PIC50s may display a DEFIB COMM ERROR, which may prevent or unacceptably delay the delivery […]
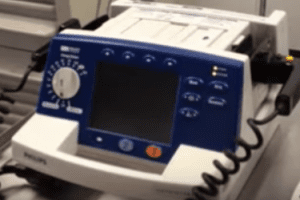
Voluntary worldwide recall of 1,184 PIC50 External Monitor/Defibrillators. MRL, Inc., a Welch Allyn Company, today announced it is initiating a voluntary worldwide recall of 1,184 PIC50 External Monitor/Defibrillators manufactured in Buffalo Grove, IL between February 2002 and October of 2004.
These PIC50s may display a DEFIB COMM ERROR, which may prevent or unacceptably delay the delivery of therapy, which may fail to resuscitate the patient. This problem occurs because of an intermittent electrical connection within the device.
The company has received 18 related complaints about devices in this group of PIC50s, corresponding to 1.5 percent of the 1,184 recalled devices, which the company deems an unacceptable risk. In two instances the “Defib Comm Error” delayed patient resuscitation. The company has taken corrective action and no other devices besides those manufactured between the above dates are subject to this recall.
Most of the Defib Comm errors are detected by a self test that is automatically performed when the PIC50 is powered up, and many of the errors are transient. If the Defib Comm error is detected by the self test, the device should be serviced before further use on patients to eliminate the risk that the problem might recur when defibrillation of a patient is required.
MRL, Inc. initiated notification via certified mail on June 30, 2006 to its customers who purchased PIC50s in this group of devices, 673 of which were sold within the US and 511 outside of the US. Owners of PIC50s in the affected population should contact MRL, Inc. using the response form included with the recall notification they receive by registered mail.
MRL will schedule customers’ returns of PIC50s for service, giving priority to the units that have displayed the Defib Comm error. Customers will be provided with loaner units at no charge while their units are being serviced. MRL, Inc. will pay all costs associated with shipping, handling and replacement of the units “DEFIB” board.
This recall is being conducted with the full knowledge of the U.S. Food and Drug Administration (FDA). FDA has determined that this action is a Class I recall. The FDA defines Class I as a situation in which there is reasonable probability that the use of or exposure to the product will cause serious adverse health consequences or death.
Customers with questions may contact the company at 1.800.462.0777 or 1.847.520.0300 for more information. Any adverse reactions experienced with the use of this product, and/or quality problems should also be reported to the FDA’s MedWatch Program by phone at 1-800-FDA-1088, by Fax at 1-800-FDA-0178, by mail at MedWatch, HF-2, FDA, 5600 Fishers Lane, Rockville, MD 20852-9787, or on the MedWatch website at www.fda.gov/medwatch.
The personal injury attorneys at Parker Waichman offer free, no-obligation case evaluations. For more information, fill out our online form or call 1-800-YOURLAWYER (1-800-968-7529).