
Excellent service from the best lawyers in New York Their customer service is excellent Especially Julie, Maryann, Dawn and the hardest the best JORGE Thank you very much to all
Jhony Gonzalez
6 months ago
St. Jude Defibrillator Side Effects. In November 2007, reports emerged that St. Jude Riata Defibrillator Lead wires had perforated the hearts of some patients. That month, the medical journal Pace published a report detailing four instances in which the St. Jude Riata Defibrillator Lead wire detached and perforated the heart wall. In one instance, the […]
St. Jude Defibrillator Side Effects. In November 2007, reports emerged that St. Jude Riata Defibrillator Lead wires had perforated the hearts of some patients. That month, the medical journal Pace published a report detailing four instances in which the St. Jude Riata Defibrillator Lead wire detached and perforated the heart wall.
In one instance, the defective St. Jude Riata Defibrillator Lead wire not only perforated the heart, but it nearly poked through a patient’s skin.
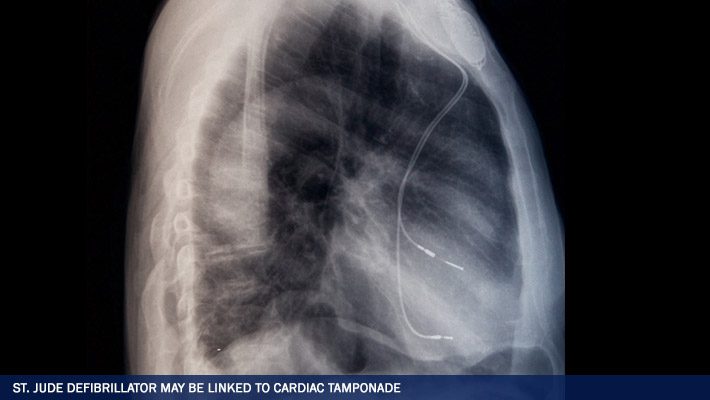
When a defibrillator lead wire becomes detached from the heart, the device will fail to emit needed electrical shocks to the heart, leaving a patient at risk of death. If the detached St. Jude Riata Defibrillator Lead perforates the heart tissue, the heart may bleed into the pericardial sac surrounding the heart.
This causes a lethal condition called cardiac tamponade in which pressure builds around the heart, preventing the heart from beating effectively. In an editorial accompanying the Pace; article, Dr. Stephen Vlay, a cardiologist for Stony Brook University, wrote that the problems with the St. Jude Riata Defibrillator Leads could be due to an inherent design flaw, at least in some models of the Riata lead.
Earlier in 2007, the medical journal Heart Rhythm reported that the St. Jude Riata Defibrillator Lead had a higher perforation rate than what had been stated by the manufacturer. According to that article, doctors at Massachusetts General Hospital reported a perforation rate of 3.8% – or 5 out of 130 for the St. Jude Riata Defibrillator Lead.
Doctors from New York Hospital Queens also reported to Heart Rhythm that of 59 St. Jude Riata Defibrillator Leads implanted there, five had perforated. New York Hospital Queens has since stopped using the defective St. Jude Riata Defibrillator Lead.
On October 11, 2016, St. Jude Medical warned of a battery defect affecting its Implantable Cardioverter Defibrillator (ICD) and Cardiac Resynchronization Therapy Defibrillator (CRT-D) device. According to a safety alert posted on the U.S. Food and Drug Administration (FDA) website, the batteries on these heart devices may fail sooner than expected.
St. Jude is urging patients and caregivers to respond to the Elective Replacement Indicator (ERI) alert as soon as it goes off. Normally, the patient has three months to receive a new battery when the ERI alert is active.
However, St. Jude has received reports of the battery failing as early as 24 hours after receiving the ERI alert. A recall and correction were initiated.
The heart devices are used to provide pacing for patients with slow heart rhythms and administer an electrical shock in patients who have a dangerously fast heart rhythm.
The Implantable Cardioverter Defibrillator (ICD) and Cardiac Resynchronization Therapy Defibrillator (CRT-D) are implanted in the upper chest, underneath the skin and connected to the heart through “leads” or insulated wires. According to the FDA safety alert, the following ICD and CRT-D models manufactured before May 2015 are affected:
The defect stems from the devices’ lithium-based batteries. The alert states that deposits of lithium, known as “lithium clusters,” may form within the batteries of ICDs and CRT-Ds. If this occurs, the defect may disrupt the normal electrical connections and cause rapid battery failure.
In some cases, the battery fully drained a day or a few weeks after the patient received an ERI alert. This defect may be fatal; if the battery drains the device will be unable to provide a life-saving shock. “The patients most at risk are those with a high likelihood of requiring life-saving shocks and those who are pacemaker dependent,” the alert notes.
Nearly 400,000 Implantable Cardioverter Defibrillator (ICD) and Cardiac Resynchronization Therapy Defibrillator (CRT-D) devices were sold worldwide. Premature battery failure prompted 841 of these devices to be returned for analysis.
So far, the recall has been linked to two patient deaths when the battery was unable to deliver a life-saving shock. There have also been 10 reports of patients fainting and 37 reports of dizziness when the battery was unable to provide the necessary therapy.
The alert notes that these figures may be an underestimation because issues are not always reported to the manufacturer.
In April 2009, St. Jude Medical Inc., the maker of implantable devices that regulate heart rhythms, identified that it has identified a memory chip problem in a small number of some of its older devices.
While no deaths or serious injuries have been reported to the company as a result of the problem, St. Jude said in a government filing that the FDA may classify the problem as a recall.
The problem, which stems from a memory chip that St. Jude used through 2002 that is susceptible to background radiation, has not been seen in any of the devices the company currently sells. The St. Jude implantable cardioverter defibrillators (ICDs) that may be affected by the radiation problem are certain older generations of the company’s Photon DR, Photon Micro VR/DR, and Atlas VR/DR models.
So far, 60 out of an approximate 36,000 devices have been found to be affected, the company wrote in the filing. Nearly 26,000 of the devices remain in patients.
St. Jude Medical Inc. has notified doctors and federal regulators of a software problem in some models of its implantable defibrillators that could cause the heart-shocking device to malfunction. About 39,000 patients are affected by this news.
ICDs are implanted devices the size of a stopwatch that is placed in the upper chest and shock or paces an irregular heartbeat back into rhythm. St. Jude indicated that it discovered the two “anomalies” during a routine product evaluation.
In ICDs past their mid-life, a series of shocks might be skipped as the device delivers its routine of multiple shocks to revitalize the patient’s heart. The first shock would be delivered, but the device might deliver fewer than the maximum of six shocks per episode.
The devices on average last four to seven years before replacement. A second problem could cause a temporary increase in the device’s pacing rate. In addition to its shocking therapy, some patients need an ICD that can pace their heart, as well.
The affected models include: Epic DR/HF (V-233/V-337/V-338), Epic Plus DR/VR/HF(V-236/V-239/V-196/V-239T/V-239T/V-196T/V-350), Atlas DR (V-242), and Atlas Plus DR/VR/HF (V-243/V-193/V-193C/V-340/V-341/V-343).
The personal injury attorneys at Parker Waichman LLP offer free, no-obligation case evaluations. For more information, fill out our online contact form or call 1-800-YOURLAWYER (1-800-968-7529).
More battery-related cases: