
Positive: Professionalism , Quality , Responsiveness
Jessica Samantha
2 years ago
The U.S. Food and Drug Administration (FDA) just sent a warning letter to the President and Chief Operating Officer of medical device maker, NuVasive, Inc., following an inspection of the firm that took place from October 29, 2012 through November 2, 2012. An agency investigator determined that the NuVasive Affix Spinous Process Plate System is […]
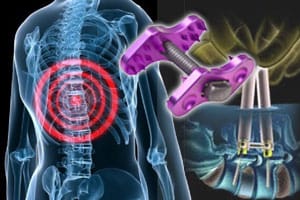 The U.S. Food and Drug Administration (FDA) just sent a warning letter to the President and Chief Operating Officer of medical device maker, NuVasive, Inc., following an inspection of the firm that took place from October 29, 2012 through November 2, 2012. An agency investigator determined that the NuVasive Affix Spinous Process Plate System is adulterated.
The U.S. Food and Drug Administration (FDA) just sent a warning letter to the President and Chief Operating Officer of medical device maker, NuVasive, Inc., following an inspection of the firm that took place from October 29, 2012 through November 2, 2012. An agency investigator determined that the NuVasive Affix Spinous Process Plate System is adulterated.
According to the agency, NuVasive did not receive pre-market approval for the device; the Affix device is misbranded; and NuVasive did not notify the agency of its intent to introduce the device into commercial distribution, which is legally mandated. Regulators warned NuVasive over promotional claims it made about these spinal products, said MassDevice.
NuVasive marketed the Affix “as an adjunct to interbody fusion (XLIF®, ALIF, PLIF, and TLIF.),” the FDA inspection report indicated, noting that, “Although your firm has multiple 510(k)s cleared for this device, none of them include the use of the Affix plate with only an intervertebral body fusion device.” Because of this, the inspectors said. “This constitutes a new intended use and a new 510(k) is required.”
FDA 510(k) clearance approvals—utilizing a so-called fast-tracked process—are permitted for medical devices that can be proven to be “substantially” equivalent to products already on the market, and don’t require human clinical trials.
NuVasive violations, wrote MassDevice, concern language about the Affix spinal device related to the NuVasive’s Interlaminar Lumbar Instrumented Fusion (ILIF) procedure and do not concern issues with manufacturing at the facility.
The Affix Spinous Process Plate System is used during surgery to clamp spinal grafts in place, FierceMedical explained. At issue, said Orthopedics This Week, is that the Affix device involved was not cleared through any of NuVasive’s Affix Spinous Process Plate system 510(k)s.
As we’ve reported previously, the 510(k) program is a frequent target of critics, who say it is too lax. In recent years, several types of medical implants approved via 510(k) protocols have been the subject of complaints, safety reviews, and lawsuits. Chief among these are metal-on-metal hip implants, including the now-recalled ASR hip implant manufactured by DePuy Orthopaedics.
Concerns about the quality of 510(k) approvals have also been raised by the growing number of serious complications linked to transvaginal mesh devices used in pelvic organ prolapse (POP) repair.