
Jill was great she took care of me the best alway call to check up on me,great law firm
Trapt Genres
6 years ago
Fourteen lawsuits against device maker Stryker allege that Stryker’s CerviCore artificial discs are so dangerous that the company abandoned a clinical trial of the device and abandoned plans to obtain premarket approval (PMA) for the product. After the fourteenth suit was filed, Judge James D. Bates of Lucas County, Ohio ordered that all 14 lawsuits […]
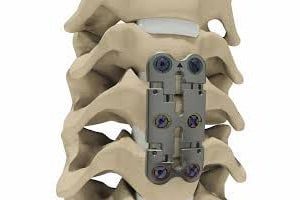 Fourteen lawsuits against device maker Stryker allege that Stryker’s CerviCore artificial discs are so dangerous that the company abandoned a clinical trial of the device and abandoned plans to obtain premarket approval (PMA) for the product.
Fourteen lawsuits against device maker Stryker allege that Stryker’s CerviCore artificial discs are so dangerous that the company abandoned a clinical trial of the device and abandoned plans to obtain premarket approval (PMA) for the product.
After the fourteenth suit was filed, Judge James D. Bates of Lucas County, Ohio ordered that all 14 lawsuits be made public, Qmed reports. The judge denied Stryker’s request to seal case documents.
Manufactures, doctors and hospitals are required to report any device-related injuries to the Food and Drug Administration (FDA). So far only two reports related to the CerviCore can be found in the FDA device database, Qmed reports. In 2008, a patient wrote, “I have severe metal poisoning, tumors and metallosis and early signs of leukemia.” The patient said the surgeon “refuses to follow up or report any of the adverse effects because it will raise red flags that he implanted a device that did not have . . . approval from the FDA.”
A recent U.S. Senate report on the failure of the FDA’s device surveillance system in a series of outbreaks of antibiotic-resistance infections linked to medical scopes, calls this “just one example of the fallacy of a system that is primarily reliant on hospitals and device manufacturers to self-report information to FDA,” according to Qmed.
The most recent CerviCore suit was brought by a Florida man who said he was injured in a clinical trial for the device. He says the device is “unreasonably dangerous,” that it had a defective design and manufacturing problems, and that he was not adequately warned about the risks. The suit claims that the CerviCore releases four times as much metal debris as Stryker initially told the FDA, and notes reports of problems with the titanium coating shearing off the CerviCore device.
The Florida man says that, unbeknownst to him, Stryker halted the CerviCore study in late 2011 and the following year stopped providing patient care related to CerviCore. The plaintiff says his health has worsened and he has possible signs of metal poisoning. The suit alleges that Stryker halted the clinical trial in part because of “horrendous manufacturing mistakes,” Qmed reports. Another patient claims the CerviCore design was defective and dislodged in her neck, causing so much nerve damage that she was forced into early retirement.
Years after the start of the clinical trial, patients began to complain of problems such as metal poisoning from the chromium, cobalt, molybdenum, and titanium debris shed by the device—similar to the problems reported by patients with metal-on-metal hip implants. Five years after the trial started, the company was still having difficulty convincing FDA to approve the device. The lawsuit claims the company sought to extend the duration of the trial from five to eight years while excluding results from patients with the most severe signs of metallosis. Further, the suit claims Stryker failed to warn patients of mounting evidence showing that the CerviCore device could cause metal poisoning and other problems.
Stryker attempted to abandon the trial but could not get the required FDA approval to do so until it agreed to disclose the known risks of metallosis. These risks, the lawsuit claims, are so severe that reasonable physicians would not use the device if they knew of the risks, according to Qmed.