
Good for the body of lawyers and all the staff, very good team, also congratulate Jorge Peniche for his excellent work, and his way of treating people, very good person.
Carlos Olmedo
5 years ago
Several models of pacemakers and implantable defibrillators should be removed. The FDA met last week with Boston Scientific/Guidant to discuss the firm’s recent announcement that several models of pacemakers and implantable defibrillators should be removed from hospital inventories because they have the potential to malfunction due to failure of the capacitor, which controls how the device […]
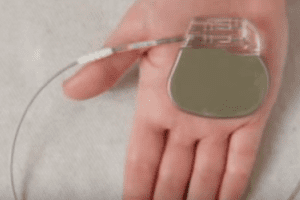
Several models of pacemakers and implantable defibrillators should be removed. The FDA met last week with Boston Scientific/Guidant to discuss the firm’s recent announcement that several models of pacemakers and implantable defibrillators should be removed from hospital inventories because they have the potential to malfunction due to failure of the capacitor, which controls how the device holds its power supply.
FDA fully supports the company’s recommendation that physicians perform follow-up exams of patients with devices that may be susceptible to this malfunction. Problems may include intermittent or permanent loss of therapy, premature battery depletion or other malfunctions.
FDA is closely monitoring the situation and met with Boston Scientific/Guidant to discuss their plans to investigation the problem, inform physicians and resolve the problem as quickly as possible.
“While information about the problem with these devices is still very preliminary, FDA is committed to keeping the public informed,” said Daniel Schultz, M.D., Director, Center for Devices and Radiological Health, FDA. “We support Guidant’s decision to notify physicians and hospitals early in the investigation, and we believe that retrieval of non-implanted devices is a prudent first step.
Analysis of returned devices may uncover clues that will allow Guidant to make further recommendations to physicians regarding patient care. Early public notification was one of the important recommendations that emerged from last fall’s meeting of the Heart Rhythm Society.”
Boston Scientific/Guidant has confirmed five reports of device malfunction among some 27,200 patients worldwide in whom these devices were implanted. One malfunction occurred at the time of implantation. In four cases, the patients needed to have the device replaced. The patients lost consciousness in two of these cases. There are no reported deaths.
Boston Scientific/Guidant has identified certain lots of a supplier’s low-voltage capacitor as the likely source of the problem. The affected products include certain models of the Insignia and Nexus pacemakers, Contak Renewal TR/TR2 cardiac resynchronization pacemakers, and Ventak Prizm 2, Vitality and Vitality 2 implantable cardioverter defibrillators.
These devices were manufactured by the company’s Cardiac Rhythm Management Group, formerly Boston Scientific/Guidant’s CRM business, and implanted in patients between December 2005 and June 2006. For more information, a letter to both patients and physicians regarding this notification is available at guidant.com/physician_communications.
Boston Scientific/Guidant is actively investigating the capacitor failure to determine the cause, the rate of occurrence, and the time to failure. Over the next 60 days, the company’s engineers will be testing the retrieved devices and analyzing the results. The company will communicate its findings to the FDA and physicians as soon as they are available.
In the meantime, as a cautionary step, Boston Scientific/Guidant has notified physicians to schedule appointments with patients implanted with affected devices as soon as possible. Physicians should examine the patient to identify any problems and to obtain information concerning when and under what circumstances these devices may fail.
“We encourage patients who have been contacted by their physicians because they may have one of the affected devices to follow their recommendation and make an appointment to have the device checked as soon as possible,” Dr. Schultz said.
“The Heart Rhythm Society applauds this proactive, collaborative effort to inform patients and physicians about the most recent device notifications. This is a monumental step in providing optimal patient care and is based on the Society’s draft recommendations released in April,” said Anne B. Curtis, M.D., FHRS, Immediate Past President, Heart Rhythm Society. “We believe the most important step is for patients to contact their heart rhythm specialist to determine the best course of action.”
FDA is interested in receiving information about problems with any devices and encourages hospitals and the public to submit reports directly to MedWatch, the FDA’s voluntary reporting program. Reports may also be submitted by phone at 1-800-FDA-1088; by fax at 1-800-FDA-0178; by mail to MedWatch, Food and Drug Administration, 5600 Fishers Lane, Rockville, MD 20857-9787; or online at http://www.fda.gov/medwatch/report.htm.
The personal injury attorneys at Parker Waichman offer free, no-obligation case evaluations. For more information, fill out our online form or call 1-800-YOURLAWYER (1-800-968-7529).