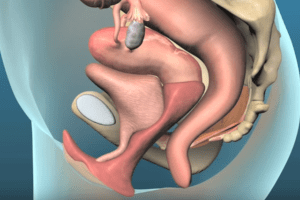
Transvaginal Mesh Recall. Parker Waichman LLP is adding its voice to those pushing for a recall of transvaginal mesh devices used in pelvic organ prolapse (POP) surgery. In a statement released yesterday, the national law firm also voiced support for a U.S. Food & Drug Administration (FDA) proposal to reclassify transvaginal mesh for POP surgery as a high-risk medical device.
“In light of the staggering number of serious complications reported among women who have received transvaginal mesh devices for POP repair, Parker Waichman LLP urges the FDA to implement its proposal to reclassify these devices as soon as possible,” the statement said.
“Parker Waichman LLP also calls on the FDA to order a recall of all transvaginal mesh devices currently being marketed for POP repair until their safety and effectiveness can be established.”
Transvaginal mesh devices approved for POP surgery
Transvaginal mesh devices were approved for POP surgery under the FDA’s 510(k) approval process. This clearance protocol is reserved for “moderate risk” devices that are substantially equivalent to devices that have already been brought to market. 510(k) clearances do not require that medical devices undergo clinical trials involving humans be conducted before they are approved.
In July, the FDA said in a safety communication that it had received 1,503 reports of serious complications associated with transvaginal mesh used in POP repair, including mesh erosion through the vagina, pain, infection, bleeding, pain during sexual intercourse, organ perforation, and urinary problems. The agency also said that its review had not seen any evidence that POP repair with transvaginal mesh offers any additional benefits compared to other treatment methods.
Highest risk category, Class III
Earlier this month, FDA staff released an Executive Summary proposing that transvaginal mesh devices for POP surgery be reclassified and placed in the agency’s highest risk category, Class III. This would mean that manufacturers could not utilize the 510(k) process, and would need to conduct human tests before applying to the FDA for approval.
The same report also proposed requiring the makers of currently approved devices to conduct postmarketing studies to assess their safety and effectiveness in POP surgery. However, the agency report did not propose recalling these devices while the studies are conducted, so they would be allowed to remain on the market.
Obstetrics and Gynecology Devices Advisory Panel
During a meeting last Thursday and Friday, in spite of the absence of a formal vote, a majority on the FDA’s Obstetrics and Gynecology Devices Advisory Panel backed the FDA proposals. While the agency is not required to follow the advice of its advisory panels, it does so in most cases.
Parker Waichman LLP is not alone in calling for a recall of transvaginal mesh devices. Some of those testifying at last week’s advisory panel meeting also called for a ban on the devices. In August, the prominent consumer advocacy group Public Citizen filed a petition with the FDA calling for such a recall, asserting that transvaginal mesh devices “offer no significant benefits but expose patients to serious risks and the potential for permanent life-altering harm.”
Need Legal Help Regarding Transvaginal Mesh Recall?
The personal injury attorneys at Parker Waichman LLP offer free, no-obligation case evaluations. For more information, fill out our online contact form or call 1-800-YOURLAWYER (1-800-968-7529).
