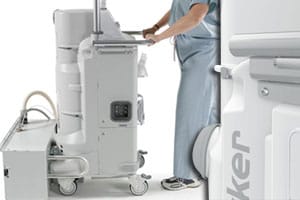
Following recalls and a U.S. Food and Drug Administration (FDA) warning letter, the agency has noted more death and injury reports associated with the Stryker Neptune Medical Waste Management System in clinics that have not stopped using the device.
The Neptune Waste Management System is meant to collect and dispose of surgical fluid waste from the patient during procedures in operating rooms and surgical facilities. In September, Stryker said it was expanding a recall it implemented for some specific Neptune Waste Management System models. At that time, Stryker noted that the devices never received mandated FDA clearance.
Stryker Corp. was forced to issue a recall on three models of its Neptune Waste Management Systems after receiving two reports of malfunctions that caused two serious patient injuries, which included one death. In one of the incidents, a patient’s passive chest drainage tube was connected to the Neptune 2 system, a model that features a strong vacuum and high flow. The patient’s death followed surgical complications caused by the malfunctioning Neptune device.
The Class I designation and recall expansion updated a prior Stryker announcement concerning defects and malfunctions with the Neptune systems. According to a previous Bloomberg News report, the initial Stryker communication indicated “that devices should not be connected to passive drainage tubes,” and that the warning was not included on then-current device safety labeling. In the updated recall statement from Stryker, customers were only first told that the FDA had never approved the Neptune 1 Silver and two Neptune 2 Ultra versions for use. At that time, Stryker admitted that the FDA did not consider the Neptune waste removal systems to be safe or effective because the agency had not approved the system for use during surgeries.
“Stryker and the FDA are particularly concerned about these cases, since a requirement for continuing to use these devices under the is to ensure that all users are adequately trained,” the FDA reported, said MassDevice. “When used incorrectly, the Neptune 1 Silver and the Neptune 2 Ultra Waste Management Systems can cause hemorrhaging and soft tissue, muscle, and vital organ damage that can lead to serious injury and/or death,” the FDA said.
Although the June warning advised customers against using the Neptune systems with high-powered surgical sucking systems when a patient suffered tissue damage and died, said Mass Device, some healthcare providers continued to use the Neptune devices. These providers submitted a “Certificate of Medical Necessity” (CMN). The providers, said MassDevice, accepted the device’s risks, agreeing to appropriately warn and train users on the device. The CMN also mandated that clinics acknowledge that “FDA does not consider the Neptune Silver or the Neptune 2 to be legally marketed devices because their safety and effectiveness have not yet been determined,” and further “certify that all users of the Neptune Silver and/or Neptune 2 at this medical facility have been informed of the contents of this notification and adequately instructed in the risk and benefits of this device and its proper use,” according to the MassDevice report.
To continue using the recalled Neptune systems, said the agency, the clinics were required to take additional steps and agree to phase out the recalled devices by March 1, 2014. At that time, said MassDevice, Stryker will stop providing all support for the recalled systems.
As we’ve explained, the Neptune Waste Management System, without mandated 510(k) clearance. Stryker noted that the FDA letter noted that Stryker Instruments submitted corrective action plans for the quality system and recall observations and that it is fully cooperating with the FDA to resolve these matters. Medical devices approved through a fast-track process known as the 510(k) do not require the formal safety and efficacy review that involves human trials. Approval is, rather, obtained by proving the device is similar to an already-approved product. Because the 510(k) route has been used to gain clearance for other controversial products, such as metal-on-metal hip implants and transvaginal mesh devices, the process has drawn increasing criticism.
Due to a lack of required approval, Stryker ceased marketing and distributing the Neptune devices. An alternative device should be sought and, if that’s not available, a certificate of medical necessity must be obtained from the FDA to continue using the device despite the lack of an approval.
