




Founding Partner
More than a decade ago, the defective medical device lawyers at Parker Waichman LLP called for increased governmental oversight of medical devices and diagnostic machines. We observed that numerous medical devices, such as two different versions of Stryker’s hip implants, heart defibrillators manufactured by Medtronic, and certain Composix Kugel Mesh patches, manufactured by C.R. Bard subsidiary Davol Inc., had caused thousands of people to suffer serious and even life-threatening injuries.
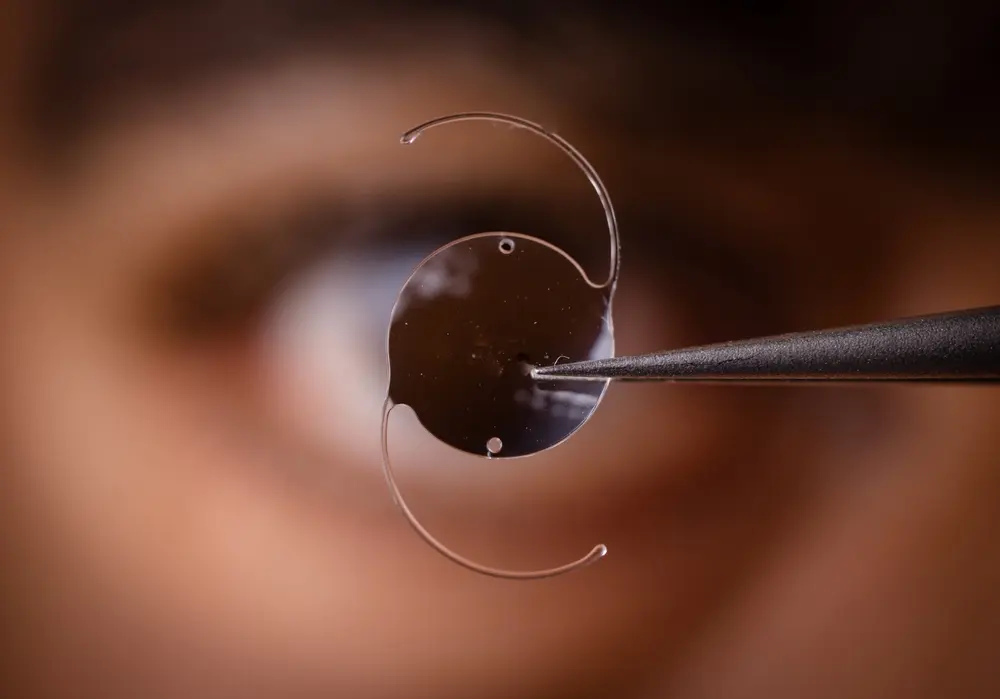
Since then, many other medical devices and treatments have failed, causing numerous people to suffer. Medical devices such as DePuy’s metal-on-metal hip implants, Bayer’s Essure birth control device, and Medtronic’s Infusion insulin pump have been linked to severe complications. Many of such devices have been recalled by the U.S. Food and Drug Administration (FDA).
Today, new FDA regulations should help to address these issues, but that won’t protect those who have already suffered injuries due to defective medical devices. If you have been harmed by a device that was supposed to help improve your health, call us. We can provide a free case evaluation to determine if you should pursue a legal claim for compensation.
In 2008, researchers from the University of California at San Francisco argued that medical devices did not have sufficiently stringent regulations to guarantee patient safety. These researchers found that new medications went through more rigorous testing and analysis before receiving FDA approval when contrasted with medical devices. (For clarity, a medical device is an implement that can be implanted, such as a knee or hip, or a machine that somehow assists the body to perform its natural functions.)
Medical device manufacturers can rely on expedited approval for their latest device when the item functions similarly to a device previously approved by the FDA. The FDA dispenses with rigorous review in these cases and permits the items to go to market. The researchers proposed that the FDA approval should be the first step toward the lawful use of medical devices, not the last step.
The next step, they suggested, should be a review of the device’s technology by an independent body of researchers and scientists. Further evaluation after the FDA’s initial approval would allow physicians to learn about potential limitations of the device and have a thorough understanding of the possible side effects from use of the device. It would also determine how long the device will last before replacement is required.
The FDA has been working on changes to its processes in recent years, but it still has a ways to go to update how it approves these potentially life-saving or life-destroying devices.
Parker Waichman LLP has offices nationwide, making it easy to access top-tier legal help no matter where you are. Our local teams are familiar with regional laws and courts, giving you a strategic advantage.
Ready to discuss your product liability case? Contact our nearest office today for a free consultation. Call now (516) 466-6500 or schedule an appointment online for a free consultation.
Parker Waichman LLP
 Amputation Injury
Amputation Injury Aviation Accidents
Aviation Accidents Birth Injury
Birth Injury Boat Accidents
Boat Accidents Burn Injury
Burn Injury Catastrophic Injury Lawyer
Catastrophic Injury Lawyer Clergy Sexual Abuse
Clergy Sexual Abuse Construction Accidents
Construction Accidents DUI Injury
DUI Injury Distracted Driving Accidents
Distracted Driving Accidents Hit-and-Run Accidents
Hit-and-Run Accidents Lyft Accidents
Lyft Accidents Nursing Home Abuse
Nursing Home Abuse Pain and Suffering
Pain and Suffering Rideshare Accidents
Rideshare Accidents Class Action Lawsuits
Class Action Lawsuits Baby Injury Lawsuits
Baby Injury Lawsuits Toxic Torts
Toxic Torts Defective Products
Defective Products Defective Medical Devices
Defective Medical Devices Defective Drugs
Sexual Abuse
Defective Drugs
Sexual Abuse  9-11 Victim Compensation
Traumatic Brain Injuries
9-11 Victim Compensation
Traumatic Brain Injuries Labor Law Injuries
Labor Law Injuries Scooter Accidents
Scooter Accidents Spinal Cord Injuries
Workers' Compensation
Spinal Cord Injuries
Workers' Compensation Intentional Acts
Intentional ActsWhy Trust UsWith Your Case?
We Take Care of Everything
No Recovery = No Fees
Decades of Experience
Recovered Billions of Dollars
Respected by Our Peers
Amputation Injury
Aviation Accidents
Birth Injury
Boat Accidents
Burn Injury
Catastrophic Injury Lawyer
Clergy Sexual Abuse
Construction Accidents
DUI Injury
Distracted Driving Accidents
Hit-and-Run Accidents
Lyft Accidents
Nursing Home Abuse
Pain and Suffering
Rideshare Accidents
Class Action Lawsuits
Baby Injury Lawsuits
Toxic Torts
Defective Products
Defective Medical Devices
Defective Drugs
Sexual Abuse
9-11 Victim Compensation
Traumatic Brain Injuries
Labor Law Injuries
Scooter Accidents
Spinal Cord Injuries
Workers' Compensation
Intentional ActsIf you or a loved one has been injured in an accident or have been injured by another party in some other way, we are here to stand up for your rights. Our personal injury attorneys have been representing injury victims and their families in Long Island and throughout the nation since the early 1980s.
In 2019, new global compliance requirements took effect that added risk management to every element of a medical device manufacturer’s quality assessment systems according to ISO 13485. The International Organization for Standardization’s guidelines are used worldwide as a uniform set of standards governing the use of many manufacturing industries, including the making of medical devices.
ISO 13485 was updated to incorporate the latest technological advances as well as regulatory mandates and quality management obligations. The upgrades to ISO 13485 will require medical device manufacturers to incorporate risk-management best practices into every aspect of the manufacturing process as well as the overall supply chain.
While adherence to ISO compliance regulations is voluntary, compliance with FDA and European manufacturing regulations is not. More stringent requirements imposed by amended FDA regulations focus on device companies showing that their new product not only is safe for use by patients, but can efficiently treat the condition for which the manufacturer designed the product.
One challenge to this move is that some device manufacturers maintain antiquated systems that make it more difficult to expose their documentation to public oversight. There is a growing need to increase the visibility of documents, results of clinical trials, and follow-up analyses, and companies will need to invest in newer technologies so that they can comply with regulatory mandates.
Whether the new regulations will make medical devices safer and help patients combat the physical ailments from which they suffer remains to be seen, but it would appear to at least be a step in the right direction.
Parker Waichman LLP
4.8 from 549 Reviews
Our law firm is ready to represent you in your injury case. We’ve helped many New York residents as well as those needing help nationwide. Contact our team for a free case consultation today.
I had a great experience with them , everyone was very helpful and sweet.
Michelle Murphy
4 months ago
Zarahi was very professional and very Quick and very knowledgeable i realy appreciated her patience and perseverance she Deserves 100 stars 🌟 but since i can only send 5 i Guess i will just have to send that truly yours Rashine Downs
Kush Three
6 months ago
VERY NICE WORK PLACE THEY HAVE BEEN GOOD TO MY MOM
Whitney Brinson
5 years ago
They treated me with tender love and care
Terrell Weaver
2 months ago
Wonderful people. They made the whole process if dealing with a gov’t agency so easy. Special compliments to Gina Viti
Michael Ross
2 months ago
I’m a 9/11 first responder, and I can honestly say that Parker Waichman made me feel like they had my best interest in my VCF case. Ms Candalino & Ms Viti are top notch in my book. I was constantly informed on the status of my case. I would definitely recommend Parker Waichman LLP to family and friends.
D D
6 years ago
We have the experience and the skilled litigators to win your case. Contact us and speak with a real attorney who can help you.
We handle mass torts cases nationwide. Please contact our office to learn more.




